

Case Study: Transforming Agriculture with AI-Engineered AgriBoost Enzyme
Download

CD Biosynsis is a leading expert in the field of enzyme design and evolution, with a long and in-depth experience in enzyme directed evolution. Our dedicated team utilizes evolutionary trace analysis to extract key information from enzyme sequences, providing valuable insights for the rational design of enzymes with improved properties or novel functions.
Evolutionary trace is a method for identifying functionally important residues in proteins. It closely links the sequence variation to the evolutionary differences, as the differences between different sequences are matched to their functional differences. In other words, evolutionary trace is actually a quantitative test for sequence differences versus functional differences. The functionally important residues in proteins are often conserved during evolution, so we are able to leverage the evolutionary trace method, which allows us to analyze a diverse set of related protein sequences to identify their conserved sequences and predict the functional significance of specific residues.
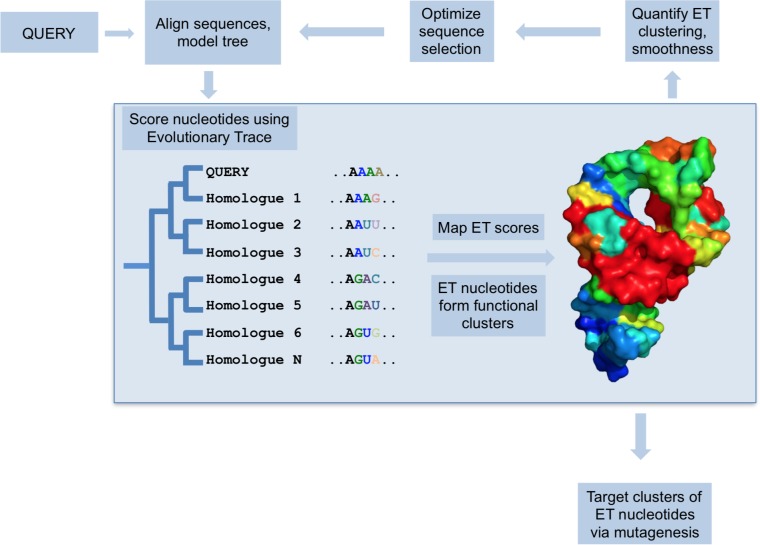 Fig 1. The evolutionary trace model. (Novikov IB, et al., 2020)
Fig 1. The evolutionary trace model. (Novikov IB, et al., 2020)
Based on the evolutionary trace method, our EnzymoGenius™ platform provides prediction and identification services of active sites or key catalytic amino acid residues of enzymes, enabling researchers to better carry out enzyme engineering studies and applications.
CD Biosynsis strives to offer comprehensive and reliable enzyme evolution trace analysis for various applications, whether academic research, industrial applications, or biotechnological development, our services are tailored to meet your specific requirements. Contact us today to learn more about our enzyme evolutionary trace analysis services and how they can benefit your research or development projects. Our team is ready to assist you and provide the insights you need to achieve your goals.
Reference
Please take a moment to fill out the form.