

Case Study: Spice up the Food and Beverage Industry with AI-Engineered Fermentation Enzyme
Download

Protein-Ligand Interaction Modeling is a crucial aspect of computational biology, with far-reaching implications in the world of medicine and scientific research. By studying and understanding the interactions between proteins (complex molecules that carry out various functions within living organisms) and ligands (molecules that bind to these proteins), we can gain insights into how these interactions influence biological processes. This understanding plays a significant role in drug design and discovery. With the knowledge of these interactions, researchers can design drugs (ligands) that bind effectively to their target proteins, potentially leading to the development of new therapeutic drugs that can revolutionize the treatment of various diseases.
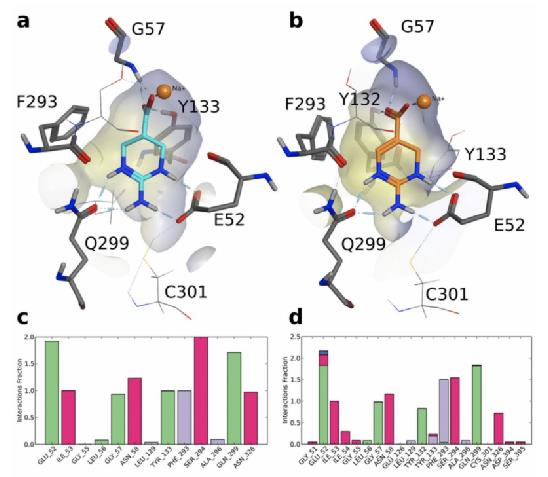 Protein-Ligand Interaction Modeling (Stefanie Kickinger, et al., 2020)
Protein-Ligand Interaction Modeling (Stefanie Kickinger, et al., 2020)
Our process for Protein-Ligand Interaction Modeling is comprehensive and harnesses advanced computational techniques and software. It can be broken down into the following steps:
| Steps | Objective | Description |
|---|---|---|
| Molecular Docking | Predict optimal ligand orientation | Initiates the process by anticipating the optimal orientation of a ligand when it binds to its target protein. This step provides a preliminary understanding of the interaction between the two molecules, laying the foundation for subsequent analysis. |
| Understanding Interactions | Explore protein-ligand interplay | Delve deeper into the nature of protein-ligand interactions, analyzing how each molecule affects the other. This step involves a detailed examination of docking results, enhancing our understanding of the molecular interaction and preparing for further investigation. |
| Molecular Dynamics Simulations | Visualize dynamic movements and interactions | Conduct molecular dynamics simulations to observe the microscopic movements and interactions of the protein-ligand complex. This dynamic view provides a more realistic portrayal of the biological process, offering insights into the behavior of the molecules in a dynamic environment. |
| Analysis of Molecular Dynamics | Examine trajectory and interaction evolution | Analyze the results of molecular dynamics simulations, focusing on the trajectory of the ligand and its interaction with the protein over time. This analysis reveals how the interaction evolves, providing crucial insights into the behavior of the molecules in a biological context. |
| Free Energy Calculation Methods | Assess stability and feasibility | Utilize free energy calculation methods to evaluate the stability and feasibility of the protein-ligand interaction. These calculations provide a quantitative measure of the likelihood of the interaction, aiding in assessing the potential effectiveness of a drug. |
| Integration of Results | Combine insights from docking, simulations, etc. | Integrate insights gained from molecular docking, dynamics simulations, and energy calculations to create a comprehensive model of the protein-ligand interaction. This holistic approach ensures a thorough understanding of the interaction dynamics and contributes to accurate modeling. |
| Validation of Model | Confirm model accuracy | Validate the accuracy of the models through thorough validation procedures. This step ensures the reliability and usefulness of the models in drug discovery and other relevant fields, verifying their alignment with experimental data and established principles. |
| Report Generation | Compile detailed findings | Compile a comprehensive report summarizing the findings of the modeling process. This report includes all data and insights gained from molecular modeling, presented in a clear and understandable format. It serves as a valuable resource for further analysis and decision-making in drug discovery and related domains. |
Here's a detailed look at our five-step process for our Protein-Ligand Interaction Modeling service:
We're here to assist you. If you have any questions, need more information, or would like to discuss a potential project, please don't hesitate to contact us. Our team is always eager to help and share our expertise.
| Application | Description |
|---|---|
| Drug Discovery | Utilizes protein-ligand interaction modeling to expedite drug discovery processes. This approach predicts and analyzes how potential drug molecules interact with target proteins, aiding in the identification of promising drug candidates and optimization of drug efficacy. |
| Enzyme Engineering | Applies protein-ligand interaction modeling in enzyme engineering endeavors. This involves predicting and understanding how enzymes interact with ligands, guiding the design of enzymes with tailored properties for various biotechnological applications, including biocatalysis and metabolic engineering. |
| Structure-Based Drug Design | Utilizes protein-ligand interaction modeling for structure-based drug design. This approach involves the rational design of new drugs based on the understanding of protein-ligand interactions, aiming to improve drug potency, selectivity, and safety profiles. |
| Virtual Screening | Employs protein-ligand interaction modeling in virtual screening of compound libraries. This computational approach rapidly evaluates the binding affinity of small molecules to target proteins, facilitating the identification of potential drug candidates for further experimental validation. |
| Molecular Dynamics Studies | Utilizes protein-ligand interaction modeling in molecular dynamics studies. This involves simulating the dynamic behavior of protein-ligand complexes, providing insights into their stability, flexibility, and conformational changes over time, which are crucial for understanding biological processes and drug mechanisms. |
Here's a brief overview of some commonly asked questions about Protein-Ligand Interaction Modeling, which touch on its applications, the software used, the accuracy of the models, and the potential influences on the binding affinity of a ligand to a protein. If you don't find the answer you're looking for, feel free to reach out to us.
Q: How does understanding protein-ligand interactions help in drug discovery?
A: By understanding how a potential drug (ligand) interacts with its target (protein), researchers can design drugs that bind more effectively. This can lead to the development of drugs with increased efficacy and fewer side effects.
Q: What is molecular docking?
A: Molecular docking is a computational method used to predict the preferred orientation of one molecule (ligand) to a second (protein) when bound to each other. It is a critical step in understanding protein-ligand interactions.
Q: What are molecular dynamics simulations?
A: Molecular dynamics simulations are computational methods that simulate the physical movements of atoms and molecules. These simulations are used to model and analyze the interaction between the protein and the ligand over time. They provide detailed information on the behavior and movement of the ligand within the protein's binding site, which can be crucial for understanding the stability of the interaction and the potential effects of the ligand.
Q: What is the role of free energy calculation methods in protein-ligand interaction modeling?
A: Free energy calculations are computational methods used to estimate the binding affinity between the protein and the ligand. This is a measure of how strongly the ligand binds to the protein. High binding affinity indicates a strong interaction, which is usually desirable in drug design. Understanding the binding affinity can help researchers optimize the ligand to improve its effectiveness.
Q: What software is used for protein-ligand interaction modeling?
A: Various software packages are available for this purpose, including AutoDock, Dock, Glide, MOE, and others.
Q: Can the techniques used in protein-ligand interaction modeling be applied to other types of molecular interactions?
A: Yes, these techniques can also be applied to other types of molecular interactions, such as protein-protein interactions or protein-DNA interactions.
Q: How accurate are the models generated by protein-ligand interaction modeling?
A: The accuracy of the models can vary based on the methods used, the quality of the input data, and other factors. However, with appropriate validation, these models can provide valuable insights into molecular interactions.
Q: Why do we need to perform both molecular docking and molecular dynamics simulations?
A: Molecular docking provides an initial prediction of the ligand's orientation and position within the protein's binding site, while molecular dynamics simulations allow us to observe how the interaction evolves over time, providing a more dynamic and realistic view of the interaction.
Q: What factors can influence the binding affinity of a ligand to a protein?
A: Many factors can influence binding affinity, including the size and shape of the ligand and protein, the presence of specific chemical groups, and the overall charge distribution of the molecules.
Q: Can protein-ligand interaction modeling be used to predict side effects of drugs?
A: Yes, by identifying potential off-target interactions of a drug, these models can help predict potential side effects.
Please take a moment to fill out the form.