Enzymes are a class of highly functional biocatalysts that catalyze chemical reactions through complex active sites, greatly accelerating the reaction process and providing unparalleled selectivity.
With the advancement of technologies such as DNA synthesis and sequencing, bioinformatics, and computational modeling, more and more natural enzymes with specific catalytic functions have been discovered. These enzymes have become key components in synthetic biology research, and breakthroughs have been made in the rational design of reaction routes for many target molecules through the assembly of different enzyme elements.
However, the fact is that there are very few natural enzymes that can be directly applied, regardless of the reaction conditions in cross species cellular environments or cell-free systems. Therefore, in the past, on the basis of natural enzymes, people often needed to modify them through protein engineering to optimize their practical application performance.

Top-down enzyme engineering (source: Nature)
In this process, many modifications and screening methods for “enzymes” have been developed, including directed evolution, ultra-high throughput screening, and phage assisted continuous evolution.
Although powerful, these not entirely rational design methods still require a significant investment of time and effort. In addition, for many ideal chemical reactions, in reality, there are no known natural enzymes that can be used as starting templates for evolution. And these have greatly limited the potential application of “enzymes”.
Although top-down excavation engineering of natural enzymes is undoubtedly still an effective development method, this method can only take us here. In our view, bottom-up or de novo enzyme design, which creates new catalytic centers in proteins, will provide universal solutions for enzyme design in terms of speed and range in the future
In the latest issue of Nature, researchers from the University of Manchester, the Federal Institute of Technology in Zurich and the University of Washington wrote this.

This is a review article published on June 1, 2022. Among the corresponding authors, we can see several well-known scholars in the field of enzyme computing design, including David Baker and Donald Hilbert. In this article, the researchers reviewed the key developments in “rational design of enzymes” and provided a possible future roadmap for this field.
Constructing the life system from bottom to top, the rational design of synthetic biology is not only focused on cell and gene circuits. At the molecular level, while enzymes provide standardized components for synthetic biology, they are undoubtedly heading towards a predictable and designable future.
Now, it’s time to consider how as a field, we will achieve step by step changes to achieve faster, more reliable, and more cost-effective development of widely used enzymes
01 Overview: Rational Design of Enzymes
Bottom-up and starting from scratch enzyme design is a major “ambition” for researchers. A desired workflow is to predict the structure of enzymes with the desired catalytic function through computational modeling after selecting the reaction target.
For the structure of enzymes, further subdivision can be made in design: the host scaffold required for enzyme design can be derived from naturally occurring protein scaffolds or designed from scratch; On the other hand, cofactors including natural and unnatural amino acid side chains, as well as metal ions, can serve as key functional components in their active sites.

Bottom-up enzyme design from scratch (source: Nature)
In the first part of the paper, the researchers first reviewed the research directions of three main types of enzyme rational design in the past, targeting these sub structures. These directions were: “Development of artificial metalloenzymes”, “Addition of non natural amino acids”, and “Computational design of stable transition states”.
Development of artificial metalloenzymes
Metallase refers to a type of enzyme produced by anchoring non biological cofactors (metal ions) onto protein scaffolds, which can catalyze various reactions such as redox and hydrolysis, and participate in the conversion of many important substances in life processes.
Currently, a proven and particularly versatile method for developing artificial metalloenzymes (ArM) involves anchoring pre assembled transition metals into protein scaffolds through interactions such as covalent, metal substituted, supramolecular, and coordinated interactions. Through this method, active catalysts have been developed for various reactions, including olefin metathesis and transfer hydrogenation.

The anchoring method of transition metals in artificial metalloenzymes (source: ACS Central Science)
Another proven effective method for designing metalloenzymes is to engineer and reconstruct natural metalloproteins to install new functional components that work in synergy with natural cofactors.
There are already many classic cases in this method, such as previous researchers engineering copper (CuB) and non heme iron (FeB) binding sites into myoglobin, and developing artificial enzymes that can simulate the functions of natural enzymes “heme copper oxidase” and “nitric oxide reductase”.
In addition, creating new protein scaffolds is also a major research direction for rational design of artificial metalloenzymes.
Most research in this field mainly focuses on introducing the binding sites of cofactors (metal ions, metal porphyrins) into the design α- In the spiral beam, various functional enzymes generated from this will be able to catalyze reactions such as hydrolysis, redox, and carbene transfer.

Design a protein scaffold that enables peroxidase C
45 to catalyze carbene transfer (source: PNAS)
In addition, metal binding sites are also designed at the interface of peptides or protein subunits for the assembly of advanced structures.
Add non natural amino acids
Enzyme design methods typically rely on 20 natural amino acids, which contain a narrow range of functional groups. And this limited functionality greatly limits the choice of installation to active sites in the design from scratch.
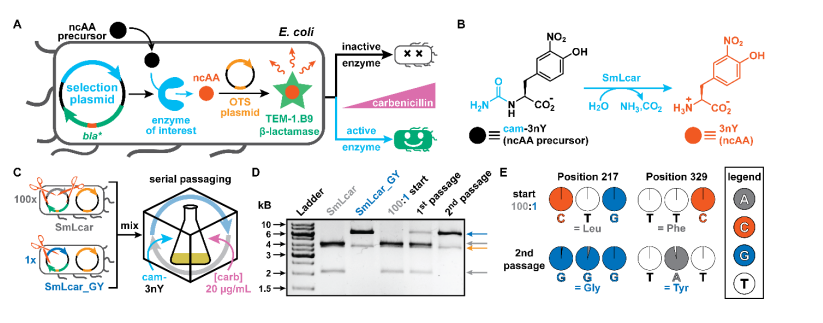
However, an extended amino acid alphabet can now be used to obtain a wider range of functional components, typically guided by orthogonal amino acid tRNA synthetase tRNA pairs, allowing for the selective addition of structurally diverse non natural amino acids (ncAA) to proteins.

Process and Example of Adding Non Natural Amino Acids (Source: Nature)
The non natural amino acids encoded by genes provide new avenues for exploring how enzymes function at the molecular level and have been used to enhance enzyme activity and stability.
Computational Design Stable Transition State
Transition state refers to an unstable high-energy state in a chemical reaction before the reactant undergoes a reaction. The reason why enzymes can catalyze reactions is that they can effectively reduce the activation energy of the reaction, and researchers have proposed the “transition state stability” theory for the underlying mechanism.
This theory suggests that the reason why enzymes reduce the activation energy of chemical reactions is because they stabilize the transition state of the substrate. Therefore, in order to design enzymes more reliably and rationally from the bottom up, the “transition state stability” in the enzyme catalytic mechanism must be studied and considered.

Enzyme catalyzed stable transition state and reduced activation energy (source: Nature)
The early screening research of “catalytic antibodies” was based on this principle.
This research is designed to use transition state analogues as antigens or haptens to stimulate the body’s immune response, resulting in the production of antibodies with catalytic properties. However, in general, antibodies cannot achieve the efficiency of natural enzymes and are difficult to handle many high energy demanding reactions.
Recently, computational enzyme design has become a more robust and flexible design method, which does not rely on limited transition state analogues and is not limited to the design of catalytic antibodies.

Computational Enzyme Design (Source: Nature Chemical Biology)
The general process of this method is as follows: 1. Calculate, design, and generate theoretical enzymes, including the key functional groups of the amino acid side chains required for stable transition states; 2. Identifying spatially complementary scaffolds by docking with proteins with structural features; 3. Redesign the residues within and around the active sites and optimize packaging.
The feasibility of this design method has been demonstrated in quite a few cases, although the initial design activity is usually low, it can be optimized through laboratory improvements. In favorable circumstances, the combination of computational design and directed evolution will enable the design and screening of functional enzymes with comparable efficiency to natural enzymes.
02 Development roadmap for rational design of enzymes
Over the past decade, we have seen significant progress in the design of enzymes from the bottom up, providing an exciting opportunity. However, if one wants to achieve or even exceed the actual utility level of top-down methods, there are two core challenges to rational enzyme design that will have to be faced and addressed directly.

Design roadmap for enzymes (source: Nature)
1. We must learn how to design highly active enzymes to make their efficiency closer to natural systems.
Efficient protein catalysis requires extremely high precision to effectively distinguish between transition and ground states, and even inaccurate positioning of side chains at the Angstrom level can have a catastrophic impact on catalytic activity. When dealing with multi-step reactions, the design challenge will also be magnified, requiring detailed conformational adjustments to accurately identify multiple chemical states.
Achieving a balance between pre organization of active sites and conformational kinetics is crucial for the future success of enzyme design.
So far, the success rate of calculating enzyme design is still low, mainly due to the limitations of the conformational sampling methods and energy functions used in existing algorithms. Although these design methods allow for rapid exploration of protein sequence space, this increased speed inevitably comes at the expense of accuracy.

Multi scale modeling design of enzymes (source: Quantum Chemistry)
To address these limitations, more complex molecular force fields are needed to accurately handle electrostatic and solvent interactions, in order to improve the accuracy of the model. Similarly, more intensive calculations, including hybrid quantum mechanics/molecular mechanics (QM/MM) methods and molecular dynamics simulations using explicit solvents, can play an important role in evaluating and improving computational design.
In addition, more effective designs can also be generated by using more complex theoretical enzyme arrangements. Although transitioning to more complex theoretical enzymes can provide more active designs, the challenge of finding suitable protein scaffolds in the recognition space increases.
The emergence of deep learning algorithms (RoseTTAFold and AlphaFold) that can accurately predict protein structure from primary sequences provides a new opportunity for designing such customized scaffolds.

David Baker is also the creator of RoseTTAFold
2. We must expand the range of chemical reactions that can be achieved by designing enzymes from scratch and develop catalysts for valuable large-scale chemical processes.
In order to maximize the effectiveness of synthesis, a particular focus that needs to be emphasized is on known non biological reaction categories that cannot be catalyzed by natural enzymes. For this situation, the design methods used in small molecule catalysis will undoubtedly be able to inspire the computational design of theoretical enzymes.
In the future, with the continuous development of the field, existing design schemes based on static models of protein transition state complexes generated from a single transition structure may become increasingly ineffective. On the contrary, a more comprehensive design method is needed to simulate multiple chemical states along the reaction coordinates to solve complex reactions involving multiple high-energy transition states.

Complex reactions with multiple transition states (source: Archives of Biochemistry and Biophysics)
In summary, although there are still considerable challenges to overcome, we are cautiously optimistic that fully programmable catalysis – the ability to predict new protein sequences from scratch to provide efficient biocatalysts with the required functions – will become a reality in the future